Skam - Skolem Assisted Makefiles
Skam is a makefile replacement. The main difference between skam and the make the utility is in how they handle pattern matching. Normal makefiles utilise text pattern matching, skam uses the even more powerful concept of function matching. These function constructs are known as skolem functions.
Unlike most makefile-type systems, skam was not developed with compiling and code management in mind. The primary reason was to support the kinds of automated computational analysis pipelines and data transformation pipelines typically required in many bioinformatics centres. However, like makefiles, skam is completely generic and should be adaptable to any task achievable with makefiles.
Skam is also built with asynchronous job submission in mind (for example, tasks involving multiple executions that must be run on a compute farm). Unlike typical makefiles, skam targets can be rows in a database as well as files on a filesystem.
Skam is written in SWI-Prolog. The full power of the prolog language is available in a skamfile specification. Prolog is particularly suited to specifying rules in a make-type system, because it is a rule-based system and has facilities for querying over facts built in. On the other hand, no knowledge of prolog is required to write skamfiles.
Example Pipeline
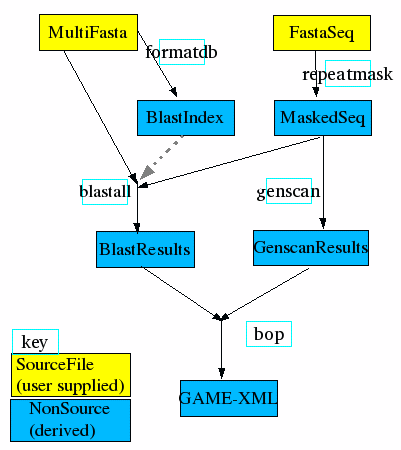
An example simple biological analysis compute pipeline. Input sequences are scanned for genes with genscan and checked for similarity to sequences from other organisms using blast. First the input sequence is masked for repetitive low-complexity sequence using RepeatMasker. Blast requires that the MultiFasta database of sequences to be compared against is first indexed using formatdb. A different blast program is run depending on whether or not the MultiFasta is of proteins or nucleic acids. Finally, the raw results are parsed and combined using the BOP parser, which exports GAME-XML.